


















 Download code from GitHub
Download code from GitHub
Loss Functions and Regularization
Loss functions are proxies that allow us to measure the error made by a machine learning model. They define the very structure of the problem to solve, and prepare the algorithm for an optimization step aimed at maximizing or minimizing the loss function. Through this process, we make sure that all our parameters are chosen in order to reduce the error as much as possible. In this chapter, we're going to discuss the fundamental loss functions and their properties. I've also included a dedicated section about the concept of regularization; regularized models are more resilient to overfitting, and can achieve results beyond the limits of a simple loss function.
In particular, we'll discuss:
- Defining loss and cost functions
- Examples of cost functions, including mean squared error and the Huber and hinge cost functions
- Regularization
- Examples of regularization, including Ridge, Lasso, ElasticNet, and early...
Defining loss and cost functions
Many machine learning problems can be expressed throughout a proxy function that measures the training error. The obvious implicit assumption is that, by reducing both training and validation errors, the accuracy increases, and the algorithm reaches its objective.
If we consider a supervised scenario (many considerations hold also for semi-supervised ones), with finite datasets X and Y:


We can define the generic loss function for a single data point as:

J is a function of the whole parameter set and must be proportional to the error between the true label and the predicted label.
A very important property of a loss function is convexity. In many real cases, this is an almost impossible condition; however, it's always useful to look for convex loss functions, because they can be easily optimized through the gradient descent method. We're going to discuss this topic in Chapter 10, Introduction...
Regularization
When a model is ill-conditioned or prone to overfitting, regularization offers some valid tools to mitigate the problems. From a mathematical viewpoint, a regularizer is a penalty added to the cost function, to impose an extra condition on the evolution of the parameters:

The parameter 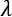 controls the strength of the regularization, which is expressed through the function
controls the strength of the regularization, which is expressed through the function 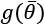 . A fundamental condition on
. A fundamental condition on 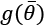 is that it must be differentiable so that the new composite cost function can still be optimized using SGD algorithms. In general, any regular function can be employed; however, we normally need a function that can contrast the indefinite growth of the parameters.
is that it must be differentiable so that the new composite cost function can still be optimized using SGD algorithms. In general, any regular function can be employed; however, we normally need a function that can contrast the indefinite growth of the parameters.
To understand the principle, let's consider the following diagram:

Interpolation with a linear curve (left) and a parabolic one (right)
In the first diagram, the model is linear and has two parameters, while in the second one, it is quadratic and has three parameters. We already...
Summary
In this chapter, we introduced the loss and cost functions, first as proxies of the expected risk, and then we detailed some common situations that can be experienced during an optimization problem. We also exposed some common cost functions, together with their main features and specific applications.
In the last part, we discussed regularization, explaining how it can mitigate the effects of overfitting and induce sparsity. In particular, the employment of Lasso can help the data scientist to perform automatic feature selection by forcing all secondary coefficients to become equal to 0.
In the next chapter, Chapter 3, Introduction to Semi-Supervised Learning, we're going to introduce semi-supervised learning, focusing our attention on the concepts of transductive and inductive learning.
Further reading
- Darwiche A., Human-Level Intelligence or Animal-Like Abilities?, Communications of the ACM, Vol. 61, 10/2018
- Crammer K., Kearns M., Wortman J., Learning from Multiple Sources, Journal of Machine Learning Research, 9/2008
- Mohri M., Rostamizadeh A., Talwalkar A., Foundations of Machine Learning, Second edition, The MIT Press, 2018
- Valiant L., A theory of the learnable, Communications of the ACM, 27, 1984
- Ng A. Y., Feature selection, L1 vs. L2 regularization, and rotational invariance, ICML, 2004
- Dube S., High Dimensional Spaces, Deep Learning and Adversarial Examples, arXiv:1801.00634 [cs.CV]
- Sra S., Nowozin S., Wright S. J. (edited by), Optimization for Machine Learning, The MIT Press, 2011
- Bonaccorso G., Machine Learning Algorithms, Second Edition, Packt, 2018






